


Services
返回Testing Services
Certification Services
- Japanese TELEC certification
- BIS certification
- PSE certification
- JATE certification in Japan
- VCCI Certification in Japan
- IMDA certification
- GCC Certification for Gulf Sev
- Iran COI certification
- Qatar COC Certification
- Iraq COC Certification
- Singapore PSB certification
- Vietnam COC certification
- KUCAS Certification in Kuwait
- KC certification
- EU E-Mark certification
- German GS certification
- REACH certification
- CE-ERP certification
- German TUV-SUD certification
- RED certification
- CE certification in Türkiye
- TSE certification in Türkiye
- Türkiye BTK certification
- European D-MARK certification
- Electrical ENEC certification
- Northern Ireland UKNI logo
- Russian EAC certification
- GOST certification in Russia
- RoHS certification
- ASTM certification
- UL, ETL, CSA, MET, TUV certifi
- FCC Certification in the Unite
- Energy Star
- IC certification
- ul certification
- ETL certification
- CEC certification
- FDA certification
- ASTM certification
- CSA certification
- CPSC Certification in the Unit
- IRAM certification
- Mexico NOM certification
- Ecuador VOC certification
- PVOC certification
- Algeria PCA certification
- Saudi EER Energy Efficiency Ce
- Saudi SABER certification
- Saudi SASO Energy Efficiency C
- Saudi EER Energy Efficiency Ce
- Iraq COC Certification
- Qatar COC Certification
- Iran COI certification
- Qatar COC Certification
- Iran COI certification
- Saudi SABER Certification Gulf
- Saudi SABER certification
- African COC certification
- Kenya PVOC certification
- Nigeria SONCAP certification
- Zimbabwe CBCA Certification
- South African LOA certificatio
- SABS certification in South Af
- Uganda PVOC certification
- Botswana COC Certification
- Zambia COC certification
- COC certification in Burundi
- Ghana COC certification
- Mauritius VOC certification
- Ethiopian VOC and COC certific
- Algeria COC certification
- Gabon COC certification
Register for the record
Systems and Training Services
Laboratory design and construction
Contact Us


National 24-hour service hotline
86+13560405821
Group Headquarters
E-mail: Lymay.zhong@lcs-cert.com
Address: Juji Industrial Park, Xueziwei, Ngabian, Shajing Street, Baoan District, Shenzhen Building A 1~2F, Building C 3F
MHRA registration in the UK
Product range:
RA registration, as a key step in the UK-bound market to ensure the continued compliance of products abroad, requires manufacturers to appoint a specialist UK regulatory person to implement, as well as prepare technical documentation in accordance with UKMDR2002, and if necessary, register with a declaration of conformity and a Notified Body certificate.
The full name of MHRA is Medicines and Healthcare products Regulatory yAgency. The MHRA is the UK's government department responsible for medicines and medical devices. After Brexit, both pre-market and post-market regulation of medical devices will be administered by the MHRA, which is similar in nature to the US FDA or the domestic NMPA.
Range of products registered by MHRA:
Prior to the Brexit transition period on January 2021, 1, medical device products registered with the MHRA mainly include:
1. Class I medical devices (including sterilization and measurement)
2. General in vitro diagnostic equipment
3. Customized equipment
After the end of the Brexit transition period on January 2021, 1, all medical devices and in vitro diagnostic medical devices need to be registered with the MHRA. Therefore, the affected are la, llb, I medical devices in MDD, and ListA and ListB devices in IVDD. MHRA registration is a new requirement for these devices.
Who carries out MHRA registration?
The MHRA only accepts applications for registration from UK manufacturers or UKResponsible Persons based in the UK. Therefore, manufacturers located outside the UK who want to complete MHRA registration must first appoint a UK Regulation Officer, and then the UK Regulation Officer will complete the registration. The concept of a person in charge of UK regulations is similar to that of the more familiar EU authorised representative.
What documents are required for MHRA registration?
1. Manufacturer information:
2. Registered address
3. Company name
4. Type of company (limited company or self-employed)
5. Contact information
6. Written agreement from the person in charge of UK regulations (where applicable)
7. Device information:
8. Applicable Regulations
9. Equipment classification
10. GMDN code of the device
11. Device name (trademark, generic name)
12. Model or version
13. Catalog number/reference number
14. UDI-DI (when applicable)
15. UK certification body (or EU notified body), applicable characteristics, such as sterilization or not, whether it contains latex, whether MRI is compatible, and the basic declaration of the category of the device, you also need to submit a conformity assessment certificate, a declaration of conformity, or a custom device declaration.
MHRA registration certification for different types of electronic cigarettes:
Medical devices and pharmaceuticals are packaged together or supplied separately
If an e-cigarette device and a nicotine-containing pharmaceutical product are separate products and the cigarette set may be reused or refilled, the cigarette device should be marked CE/UKCA as a medical device. Since e-cigarettes contain components such as batteries and heating elements, standards related to electrical components are also relevant.
Two or three e-cigarettes or one refillable e-cigarette
The MHRA believes that e-cigarette portions containing batteries and any associated charging accessories should be Class IIa active therapeutic medical devices, unless administered in a potentially hazardous manner, in which case they should be Class IIb. This means that a separate application needs to be submitted to a duly designated Notified Body/UK Approval Authority to evaluate the device elements of the e-cigarette and then mark the CE/UKCA (if applicable). Although it usually contains a heating element, cartridges containing nicotine solutions are considered part of the pharmaceutical product.
"The heating element part is still subject to the relevant essential requirements in Annex 93 of the Medical Devices Directive (42/1/EEC) applicable to the United Kingdom, or Annex 2021 of the Medical Devices Regulation applicable to Northern Ireland or the whole UK from 5 May 26 (EU 2017/745), and evidence is required to demonstrate this. It is assumed that the safety and performance of the battery case will also be checked by the notification body/UK approval authority at the same time as the inspection of the battery, as there is an intrinsic link between the two. The company needs to consider the interaction of the "device" and the heating element inside the cartridge. ”
The overall combination of pharmaceutical products and medical devices - disposable electronic cigarettes
If the device and drug product form a single, integrated product, specifically designed for a given combination, and not refillable, the entire product will be regulated as a drug and marketing authorization will be required. However, the "device" element should meet the relevant essential requirements of Annex I of the Medical Devices Directive 2002/93/EEC as amended (in the form existing as of 42 January 2002) as amended by Annex I of the Medical Devices Directive 618/2021/EEC as amended (in the form existing as of 1 January 1).
"As of 2021 May 5, the EU Medical Devices Regulation (EU 26/2017) is fully applicable to Northern Ireland and the EU." Equipment' components must meet the general safety and performance requirements for the Northern Ireland market as set out in Annex I to this Regulation and be eligible for marketing authorisation applications within the UK. In this case, there is no need to comply with the essential requirements of the Medical Devices Directive separately. These regulations also require equipment components to be evaluated by an EU notified body to demonstrate compliance with the general safety and performance requirements of the regulation, and the evaluation of an EU-designated notified body needs to be submitted with the MAA. In fact, the notified body/approval body will evaluate the equipment components, just as it also requires the CE marking/CE UK (NI) mark. ”
Nicotine-free e-cigarettes
If an e-cigarette does not contain nicotine or any other active substance, it is not considered a medicinal product and does not require marketing authorization under the definition of the Medicines for Humans Regulations 2012, as amended. If data can be provided to prove that a nicotine-free product can be used to treat a specific nicotine addiction, the product can be considered a medical device under the Medical Devices Regulation and must therefore be authorised as a medical device. Medical claims are limited to the treatment of nicotine addiction.
Electronic cigarette liquid or nicotine liquid only
1. If a medical declaration is made, only nicotine liquid can be applied for a marketing license. However, it is necessary to prove that the liquid is safe and effective in the designated e-cigarette or other vaporization device. In addition, such e-cigarette or vaporization devices need to be registered as medical devices and bear the CE/UKCA mark (if applicable).
2. As nebulizer manufacturers may change their products at any time, an agreement is required to notify nicotine liquid manufacturers/suppliers and the notified body/UK approval authority of all relevant changes to their e-cigarettes or nebulizers. Applicants for nicotine liquid MAA need to consider the possibility of liquid abuse (overdose).
Electronic cigarette MHRA registration and certification process:
1. Communicate the specific situation of the product (whether it has cigarette oil, how many flavor models)
2. Determine the cost of product testing and the number of samples
3. Sign the contract
4. Send samples and fill in the application form
5. The laboratory receives samples for testing
6. A draft report will be issued after passing the test
7. Confirm the draft report and make MHRA notification materials
8. Register an MHRA system account
9. Upload test reports and other notification materials, and pay the notification fee
10. After the notification is passed, the product will be displayed in MHRA
Note! Manufacturers of new e-cigarette and refill container products must submit a notification to the MHRA 6 months prior to their intention to place their products on the UK and/or Northern Ireland market. Once your notification is posted on MHRA's website, you can launch the product in the notification area. If the notification has been issued, you do not need to wait for the remainder of the 6 months before placing the product on the market in the notification area.
EU TPD Directive on electronic cigarette amendments:
1. The main contents of the product electronic notice should be included:
1. Manufacturer name and contact information;
2. Ingredient list and emission content;
3. Toxicological data;
4. Nicotine dose intake information;
5. Product composition and production process description;
6. Declaration of product quality and safety responsibility;
Second, the requirements of cigarette oil and liquid reservoir:
Nicotine-containing liquid is placed on the market only in professional fillers and the volume of the filled container does not exceed 10 mL, and in disposable e-cigarettes or single-use cartridges, the volume of the cartridge or liquid reservoir must not exceed 2 mL.
3. Relevant requirements for nicotine:
1. In the cigarette oil containing nicotine, the nicotine content shall not exceed 20mg/mL;
2. Under normal use conditions, the nicotine release of electronic cigarettes should be maintained at a stable level;
IV. Preparation of documents before electronic cigarette application for TPD registration:
1. Production process documents;
2. Emission test report;
3. Consistency detection method of nicotine release;
4. Refillable nebulizer open&refill instructions;
Note: Luxshare Testing can provide a complete set of testing solutions for electronic cigarettes.
V. Overview of the Registration Process (EU TPD)
1. Registration (ECAS) website: https://webgate.ec.europa.eu/cas/eim/external/register.cgi
2. Apply for a Submitter ID.
3. Fill in the form submitterid_registtatingform_en (about 10 working days to complete).
4. After the registration is completed, notify the administrator by email, stating that the ID registration is successful, and indicate the ECAS and Submitter ID.
5. After success, log in to the account opening information platform (EU-CEG) to fill in the information, upload attachments, and select different countries to submit the information separately.
6. Services that may be provided
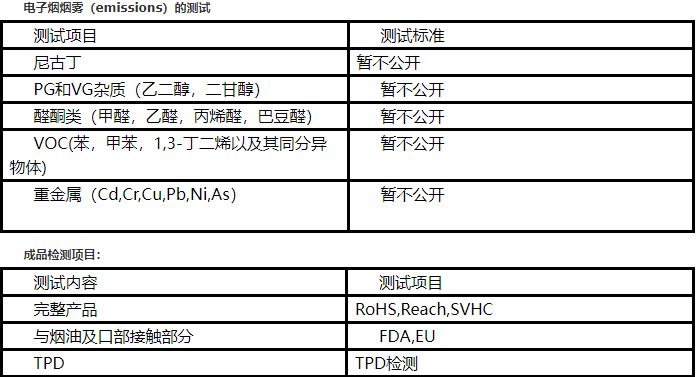
Luxshare Testing is one of the earliest independent third-party testing institutions for electronic cigarette testing in China, with a strong technical research and development team equipped with advanced testing instruments. It has a good cooperative relationship with the world's most influential e-cigarette brand. The testing method developed by Luxshare Testing for electronic cigarette oil has been recognized by the China National Accreditation Service for Conformity Assessment (CNAS), and the report issued by Luxshare has been widely recognized internationally. For the analysis of harmful components of e-cigarette emissions (Emission), we have taken the lead in developing a complete set of test programs by adopting international standards widely accepted by customers, which can provide customers with the following testing services:
1. Emission composition test for e-cigarette emissions;
2. Analysis and testing of e-cigarette oil composition;
3. E-cigarette EU registration consulting services;
4. Electronic cigarette food contact material (FCM) test;
5. Electronic cigarette finished products EU RoHS, REACH and other regulations testing.
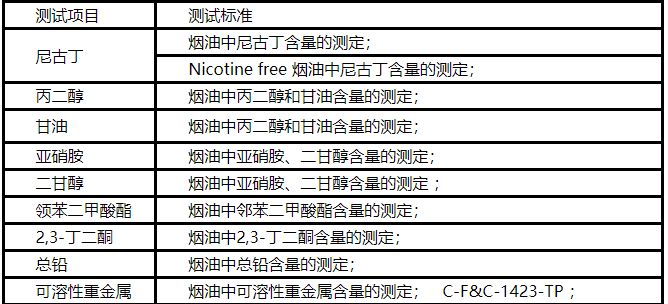
Electronic cigarette oil testing items:
Large-scale experimental base
Professional Lab
Analysis Method
Testing/year



Contact Us
National 24-hour service hotline
Juji Industrial Park, Xueziwei, Ngabian, Shajing Street, Baoan District, Shenzhen Building A 1~2F, Building C 3F

WeChat consultation
Please note consulting services
Address:Juji Industrial Park, Xueziwei, Ngabian, Shajing Street, Baoan District, Shenzhen Building A 1~2F, Building C 3F mobile:86+13560405821 Mailbox:Lymay.zhong@lcs-cert.com
Copyright © LCS Testing Lab all right reserved